

Peptide News !!!
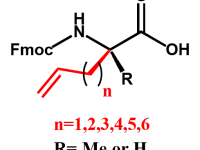



"Innovate, automate or evaporate !"

ROBERT BRUCE MERRIFIELD
Nobel Prize winning in chemistry
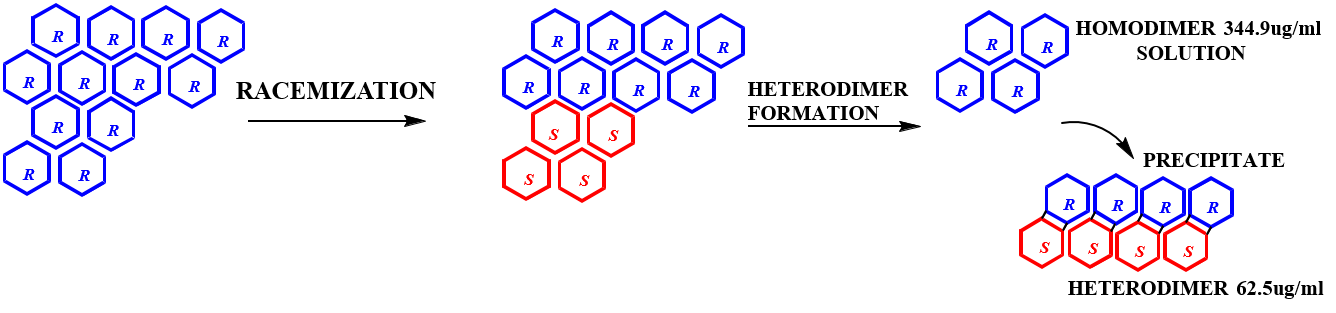
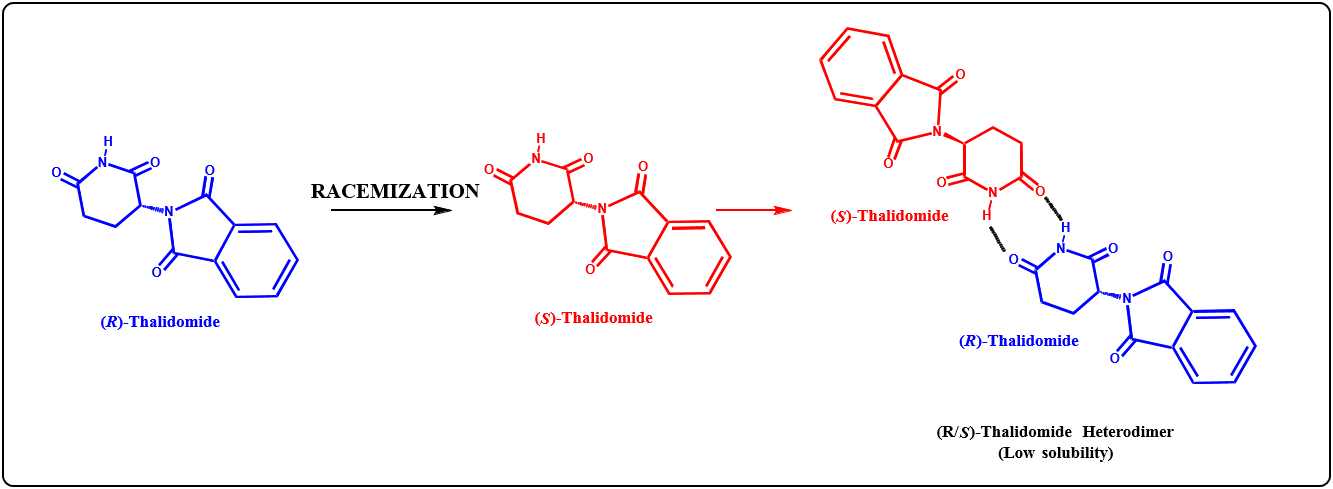

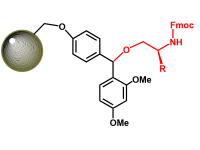
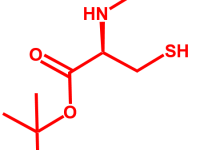
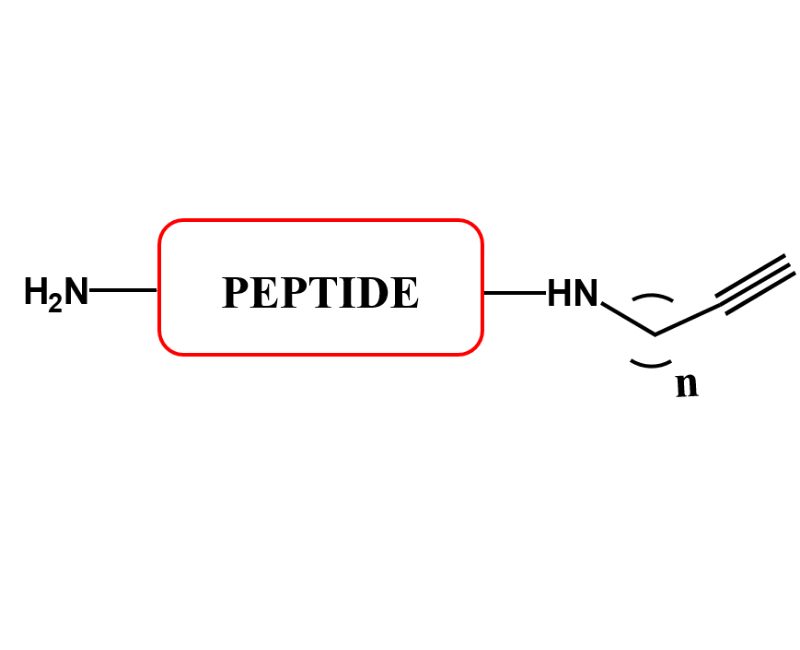
C-TERMINAL PEPTIDES
IN ADDITION TO THE ACID AND AMIDE C-TERMINI, WE CAN ALSO PROVIDE ALCOHOL, n-ALKYL AMIDES AND MORE
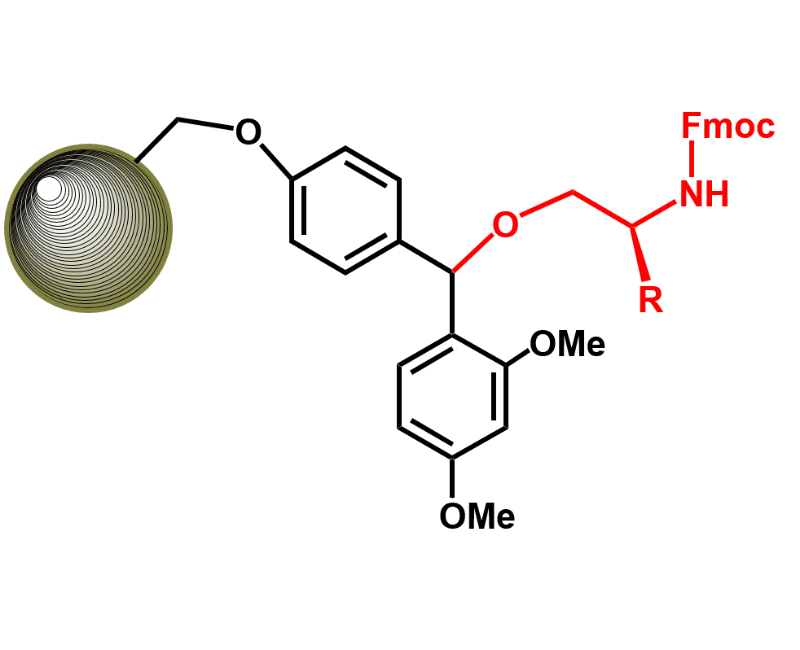
N-TERMINAL MODIFICATIONS
WE CAN PROVIDE BIOTINYLATED, PEPTIDES WITH ANY KIND OF SPACER. FLUORESCEIN, FITC AND MORE DYES. fATTY ACIDS LIKE MIRISTIC, PALMITIC AND OLEIC CAN ALSO BE INCORPORATED AL THE n-TERMINUS.
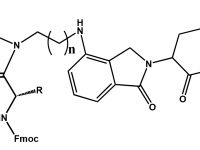
PROTAC PEPTIDE CONJUGATES
The PROTAC system for selective protein destruction. They are constructs made up of two molecules that are connected by a linker. The activity of PROTACS is dependent on these connected molecules. One end binds to the protein of interest (POI), while the other binds to an E3 ligase. PROTACs hijack the ubiquitin-proteosome system (UPS) to induce ubiquitination and degradation of the POI by bringing it in close proximity to the E3 ligase.

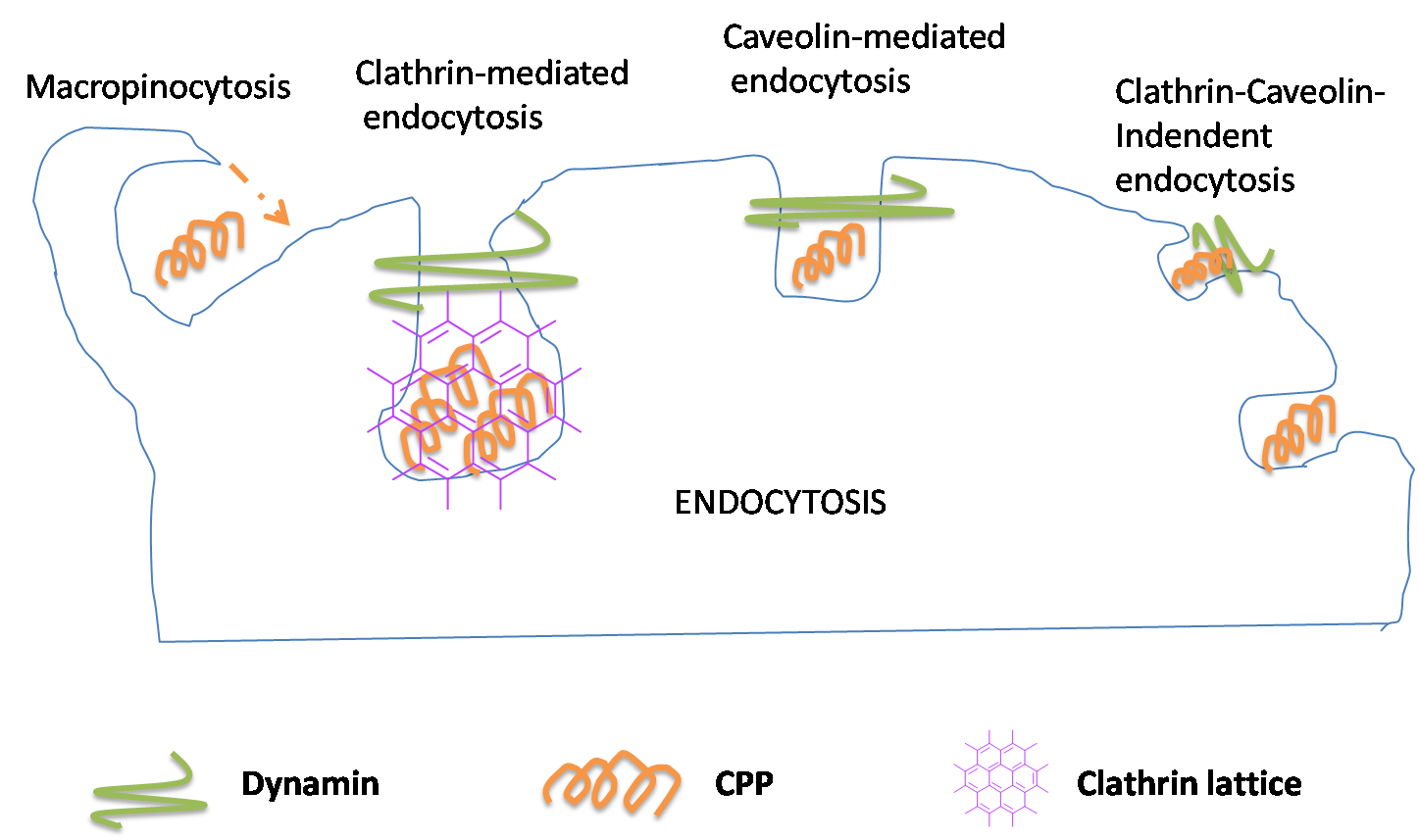
CPP PEPTIDE CONJUGATES
Cell penetrating peptides have been successfully used for delivering a wide variety of therapeutic molecules into various types of cells for the treatment of multiple diseases. These peptides can internalise in many type of cells.
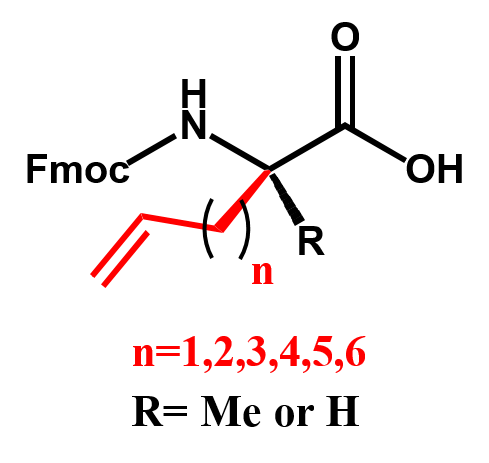
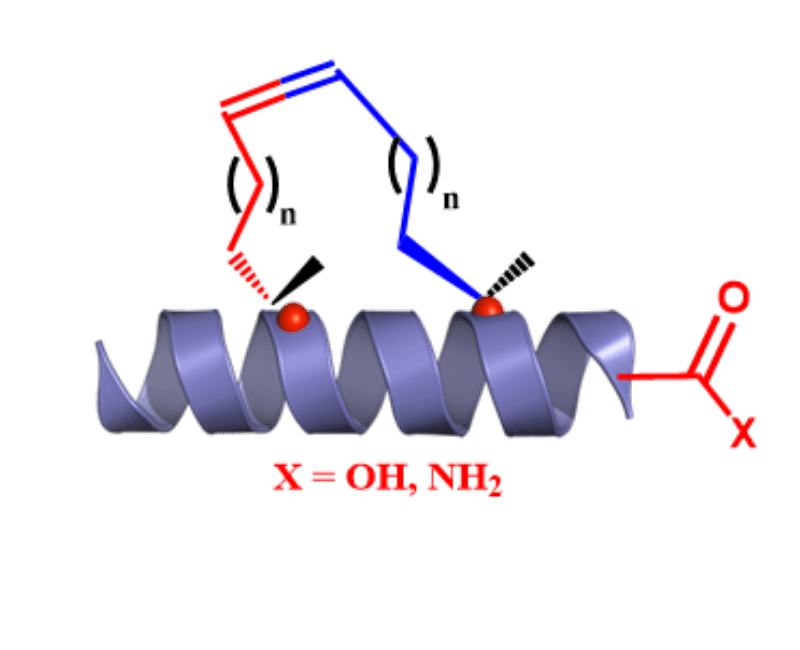
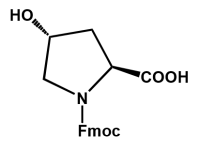
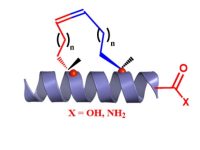
SPECIAL AMINO ACIDS
We offer competitive prices on the Fmoc-alkenyl alanines used for the synthesis of all hydrocarbon Stapled α−helical peptides (AHSP). AHSP have been used to disrupt the interaction between p53 and MDM2 and MDMX which are overexpressed in some types of cancers restoring the p53-dependent cell-cycle arrest and apoptosis in several class of tumors.

GIVE US A CALL
+1 657 242-0307 or send us an e-mail: fernando@delivertides.com
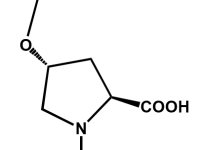
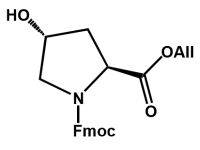

"An expert is a man who has made all the mistakes which can be made, in a narrow field".
NIELS BOHR
Nobel Price Winner in Physics